

Significance
Cardiovascular disease, most often due to atherosclerosis, is the leading cause of death both in the United States and throughout the world. During the past decades, medical therapy and life-style changes have decreased the prevalence of cardiovascular disease in the United States and other “Western” countries. However, this downward trend recently reversed in the United States, and the prevalence of cardiovascular disease is also increasing in other parts of the world. Targeted molecular interventions—that are based on a detailed understanding of the pathogenesis of cardiovascular disease—hold great promise for preventing and treating cardiovascular disease.
Overview of the Laboratory

Our projects share a common goal: to discover the mechanisms that cause cardiovascular disease and to translate these discoveries into novel therapies that prevent or reverse cardiovascular disease. There are 3 major projects:
- Development of gene-transfer vectors capable of mediating long-term recombinant gene expression in blood vessels and use of these vectors to prevent or reverse atherosclerosis in arteries and vein grafts.
- Investigation of the role of transforming growth factor β (TGF-β) signaling in the development and prevention of aortic aneurysms.
- Investigation of the role of the urokinase plasminogen activator (uPA)/plasminogen system in the development of atherosclerosis and plaque rupture; defining the molecular mechanisms of atherosclerotic plaque rupture.
Approaches, Contributions, and Current Work
We use techniques and approaches of molecular biology, biochemistry, genetics, virology, immunology, proteomics, and whole-animal physiology.
Vascular gene therapy

Since 1988, our laboratory has clarified the promise and limitations of gene-transfer vectors to express genes in the blood vessel wall.(1) We have also tested whether gene therapy can prevent or reverse vascular disease. A major advance in this work was our discovery that helper-dependent adenoviral (HDAd) vectors (which lack all viral genes) can express therapeutic genes stably in the artery wall, with minimal associated inflammation.(2) Clinical applications of this gene-therapy approach include strategies that prevent or reverse atherosclerosis in arteries and in venous bypass grafts (Figure 1). (3)

Currently, we are exploiting a rabbit model of vein-graft atherosclerosis that was developed in our laboratory. We are using this model to test HDAd-mediated gene therapy that prevents vein-graft atherosclerosis, a major clinical problem. We are also constructing and testing new transgene expression cassettes that we anticipate will drive durable high-level therapeutic transgene expression in both arteries and veins. In addition, we are developing new approaches to atheroprotective gene therapy, based on exosome-mediated transfer of inhibitory RNAs and on upregulation of the cholesterol transporter ABCA1.
TGF-β signaling in vascular disease and development

Transforming growth factor beta (TGF-beta) is a pleiotropic cytokine that is expressed in the artery wall, circulates in plasma, and plays critical roles in vascular development, homeostasis, and disease. We use mouse models of gene transfer, germ-line transgenesis, gene knockout, and antibody-mediated inhibition to uncover the roles of TGF-beta signaling in the blood vessel wall. Using these approaches, we discovered both pathogenic and protective effects of altered expression of TGF-beta and its cell-surface receptors. These effects include reduced atherosclerosis in mice with increased plasma levels of TGF-beta, and aortic aneurysm formation in mice that lack TGF-beta signaling in smooth muscle cells (Figure 2).(4) We also discovered a protective—not pathogenic—role for TGF-beta signaling in a mouse model of Marfan syndrome.(5)

Currently, we continue to use mouse models to identify the pathways through which SMC TGF-beta signaling protects the aorta and through which loss of physiologic SMC TGF-beta signaling causes aortic disease. We anticipate that insights from these experiments will clarify roles of TGF-beta signaling in vascular homeostasis, provide insights into the pathogenesis of aortic aneurysms—both genetically based and acquired—and reveal novel strategies for preserving vascular health.
The uPA/plasminogen system and vascular disease
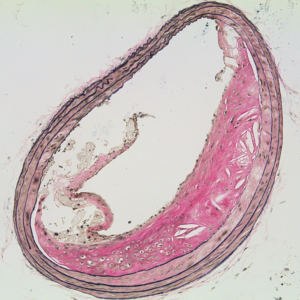
Urokinase-type plasminogen activator (uPA) is a serine protease that converts the zymogen plasminogen to plasmin (a broadly active protease). uPA circulates in plasma and is expressed by cells in the artery wall. We became interested in uPA because of its ability to act as a therapeutic agent by promoting lysis of occlusive intravascular thrombi. We first proposed using uPA (and the related enzyme tPA) for antithrombotic gene therapy delivered either directly to the vessel wall or from the surface of thrombogenic intravascular devices. We then discovered that uPA—when expressed at increased levels in the artery wall of hyperlipidemic rabbits or mice—increases atherosclerosis and causes arterial constriction. More recently, we discovered that increased expression of uPA in mouse macrophages causes atherosclerotic plaque rupture (Figure 3).(6) These results suggest that increased uPA expression in the vessel wall will not be useful for antithrombotic gene therapy. However, our experiments yielded a mouse model of plaque rupture, an important but poorly understood human disease process.
We recently discovered that ruptured plaques in uPA-overexpressing mice resemble ruptured plaques removed from human carotid arteries, both morphologically and biochemically. Currently, our work is aimed at defining the mechanisms through which uPA accelerates atherosclerosis and causes plaque rupture. Insights into the mechanisms of uPA-accelerated atherosclerosis and plaque rupture may reveal new strategies for preventing atherosclerosis and its complications.
References
-
Newman KD, Dunn PF, Owens JW, Schulick AH, Virmani R, Sukhova G, Libby P, Dichek DA. Adenovirus-mediated gene transfer into normal rabbit arteries results in prolonged vascular cell activation, inflammation, and neointimal hyperplasia. J. Clin. Invest. 1995;96:2955–2965
-
Wen S, Graf S, Massey PG, Dichek DA. Improved vascular gene transfer with a helper-dependent adenoviral vector. Circulation. 2004;110:1484-1491
-
Wacker BK, Dronadula N, Bi L, Stamatikos A, Dichek DA. Apolipoprotein a-i vascular gene therapy provides durable protection from atherosclerosis in hyperlipidemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2018;38:206–217
-
Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA. Postnatal deletion of the type II transforming growth factor-beta receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler. Thromb. Vasc. Biol. 2015;35:2647–2656
-
Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered transforming growth factor beta signaling. J Am Heart Assoc. 2017;6:pii: e004968
-
Hu JH, Du L, Chu T, Otsuka G, Dronadula N, Jaffe M, Gill SE, Parks WC, Dichek DA. Overexpression of urokinase by plaque macrophages causes histologic features of plaque rupture and increases vascular mmp activity in aged apo E-null mice. Circulation. 2010;121:1637–1644
Research leadership

Principal Investigator
David A. Dichek, MD
David A. Dichek, M.D.
Professor of Medicine
John L. Locke, Jr. Family Endowed Chair in Medicine
Associate Director for Research, Division of Cardiology
Email: ddichek@uw.edu
Contact
U.S. Mail:
1959 N.E. Pacific Street
Box 357710
Seattle, WA 98195-7710
Courier (FEDEX, UPS, etc.):
Rm K143A Health Sciences Bldg.
1959 NE Pacific St.
Seattle, WA 98195
Tel (206) 685-6959
FAX (206) 221-6346
MAKE A GIFT
Cardiovascular Research Gift Fund
Supports research into the causes of blood vessel diseases (including atherosclerosis and aneurysms) and the development of new treatments.